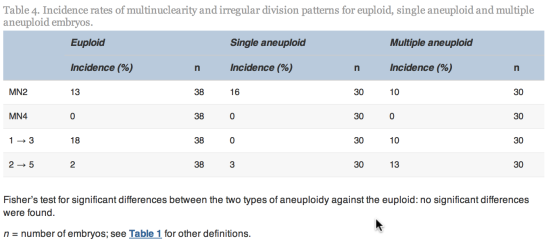
.gif)

Modelling a risk classification of aneuploidy in human embryos using non-invasive morphokineticsReproductive BioMedicine Online17 May 2013 |
Alison Campbell, Simon Fishel, Natalie Bowman, Samantha Duffy, Mark Sedler, Cristina Fontes Lindemann Hickman

Alison Campbell studied at Leicester and Nottingham Universities, specializing in assisted reproduction technology. She started her career as an embryologist in Liverpool and held a senior embryology position with CARE Fertility at its inception in 1997. Since then she has played a key role in establishing new laboratories and has headed up the embryology teams across the CARE organization. Her current role involves driving standards, best practice and leading research and development across the their laboratories in the UK and Ireland. Alison is an experienced clinical embryologist, a member of the HFEA licensed centres panel and a diplomate of the Royal College of Pathologists. Alison was responsible for the first clinical application of time-lapse microscopy in the UK.
PII: S1472-6483(13)00070-9
doi:10.1016/j.rbmo.2013.02.006
© 2013 Reproductive Healthcare Ltd. Published by Elsevier Inc All rights reserved.
This study determined whether morphokinetic variables between aneuploid and euploid embryos differ as a potential aid to select euploid embryos for transfer. Following insemination, EmbryoScope time-lapse images from 98 blastocysts were collected and analysed blinded to ploidy. The morphokinetic variables were retrospectively compared with ploidy, which was determined following trophectoderm biopsy and analysis by array comparative genomic hybridization or single-nucleotide polymorphic array. Multiple aneuploid embryos were delayed at the initiation of compaction (tSC; median 85.1 hours post insemination (hpi); P![]() =
=![]() 0.02) and the time to reach full blastocyst stage (tB; median 110.9
0.02) and the time to reach full blastocyst stage (tB; median 110.9![]() hpi, P
hpi, P![]() =
=![]() 0.01) compared with euploid embryos (tSC median 79.7 hpi, tB median 105.9 hpi). Embryos having single or multiple aneuploidy (median 103.4 hpi, P
0.01) compared with euploid embryos (tSC median 79.7 hpi, tB median 105.9 hpi). Embryos having single or multiple aneuploidy (median 103.4 hpi, P![]() =
=![]() 0.004 and 101.9 hpi, P
0.004 and 101.9 hpi, P![]() =
=![]() 0.006, respectively) had delayed initiation of blastulation compared with euploid embryos (median 95.1
0.006, respectively) had delayed initiation of blastulation compared with euploid embryos (median 95.1![]() hpi). No significant differences were observed in first or second cell-cycle length, synchrony of the second or third cell cycles, duration of blastulation, multinucleation at the 2-cell stage and irregular division patterns between euploid and aneuploid embryos. This non-invasive model for ploidy classification may be used to avoid selecting embryos with high risk of aneuploidy while selecting those with reduced risk.
hpi). No significant differences were observed in first or second cell-cycle length, synchrony of the second or third cell cycles, duration of blastulation, multinucleation at the 2-cell stage and irregular division patterns between euploid and aneuploid embryos. This non-invasive model for ploidy classification may be used to avoid selecting embryos with high risk of aneuploidy while selecting those with reduced risk.
Chromosome anomalies (aneuploidy) are prevalent in human embryos and result in either IVF failure, miscarriage or the birth of children with chromosome disorders. After IVF, it is imperative to try to detect aneuploid embryos in order to transfer an embryo with a normal chromosome copy number (euploid) to maximize the chances of a successful and healthy live birth. Currently, the only option is to biopsy a cell (or cells) from the embryo and analyse these using complex and expensive genetic technology. Novel uninterrupted culturing methods using the EmbryoScope now enables detailed observation of the each embryo via time-lapse photography during the whole culture period, for up to 6![]() days. This new technology has permitted us to observe the development of human embryos in detail after IVF, providing analysis of complex patterns of cell division and cell movement (morphokinetics). As far as is known for the first time, we have demonstrated differences between aneuploid and euploid embryos based on their morphokinetic patterns, whereby aneuploid embryos are significantly delayed in reaching the later stages of development, during days 4 and 5 in culture. We postulate that, by using the unique, non-invasive and specifically designed models or algorithms developed in this study, embryologists can make more informed choices on the most viable embryo to select for transfer and reduce the risk of selecting an aneuploid embryo.
days. This new technology has permitted us to observe the development of human embryos in detail after IVF, providing analysis of complex patterns of cell division and cell movement (morphokinetics). As far as is known for the first time, we have demonstrated differences between aneuploid and euploid embryos based on their morphokinetic patterns, whereby aneuploid embryos are significantly delayed in reaching the later stages of development, during days 4 and 5 in culture. We postulate that, by using the unique, non-invasive and specifically designed models or algorithms developed in this study, embryologists can make more informed choices on the most viable embryo to select for transfer and reduce the risk of selecting an aneuploid embryo.
Keywords: aneuploid, embryo, EmbryoScope, morphokinetics, PGS, time-lapse monitoring
Aneuploidy is prevalent in human oocytes and resulting embryos and its incidence increases with maternal age. Depending on the age of the female or the particular circumstance of the couple, more than half of their embryos are likely to be aneuploid (Fragouli and Wells, 2011). Avoiding the selection of aneuploid embryos for transfer after IVF is a clinical imperative. The only method available for assessing embryo ploidy uses invasive and expensive preimplantation genetic screening (PGS), which provides accurate full chromosome copy number of a biopsied blastomere from the early embryo, trophoblast cells of the blastocyst or the polar bodies of the oocyte and zygote. Various robust and reliable technologies have been recently introduced, such as array comparative genomic hybridization, single-nucleotide polymorphism arrays or quantitative PCR, which have confirmed the high incidence of aneuploidy in human oocytes and embryos (Fragouli et al., 2006, Fragouli et al., 2010, Kamiguchi et al., 1993, Munne et al., 1993). One of the main causal factors of aneuploidy is believed to be premature pre-division of chromatids and non-disjunction during meiosis, but several other contributory factors such as paternal or mitotic aneuploidy have been implicated. Aneuploidy in human embryos results in miscarriage, implantation failure or the birth of an affected child. PGS provides couples at high risk of embryo aneuploidy the opportunity for clinics to selectively transfer a euploid embryo, if available. Recently, it has been shown that the incidence of live birth in young, good-prognosis women, with a theoretically low risk of embryo aneuploidy is enhanced by PGS (Yang et al., 2012). Furthermore, where there is the need to transfer only a single or a maximum of two embryos, either by regulation (such as in the UK) or good practice, the selection of embryos from either euploid oocytes or embryos has improved outcome in specific circumstances (Fishel et al., 2010, Fishel et al., 2011, Potter et al., 2012, Schoolcraft et al., 2012).
The development of commercially available time-lapse devices for the IVF laboratory during recent years has enabled embryologists to observe and study the development of human embryos continuously and, therefore, more precisely than was possible previously. Such monitoring of the dynamics of embryo development, in addition to traditional qualitative morphological observations, often referred to as morphokinetics, provides a plethora of information on the development of individual embryos. These data, generated by the manual or automatic recording or annotation of images collected over precise time points, can be retrospectively analysed against outcome variables, such as blastulation, implantation, ploidy or live birth in an attempt to identify prospective selection algorithms for embryo transfer. Morphokinetic analyses promise to provide IVF practitioners with novel markers of embryo viability. Recent studies (Dal Canto et al., 2012, Meseguer et al., 2011,Meseguer et al., 2012, Pribensky et al., 2010, Wong et al., 2010) have discussed the potential value of morphokinetic variables measured by time-lapse monitoring for improved embryo selection.
The application of time-lapse embryo monitoring in a clinical IVF setting avoids the need to remove embryos from incubation conditions to make a daily observation. It allows embryologists to rewind or freeze images in order to consider the detail and context of embryo development with practical flexibility. The EmbryoScope is the first instrument to provide a safe, stable incubation environment (with low incubation volume and direct heat transfer) combined with internal microscopy. Meseguer et al. (2012) observed a 20% increase in pregnancy rate compared with standard incubation and attributed this improvement to the EmbryoScope’s stable culture conditions and use of morphokinetic variables for embryo selection. This study centre’s experience is similar and this was the reason for the EmbryoScope being selected for this study.
As both ploidy analysis from PGS and morphokinetic variables from time-lapse imaging are two techniques that, individually, have been demonstrated to have the potential to significantly improve the incidence of clinical pregnancy compared with traditional culture and selection methods, this study examined whether euploid or aneuploid embryos display differing morphokinetic variables over the preimplantation cleavage period. Using trophectoderm biopsy subsequent to controlled culture conditions, the objective of this study was to determine whether embryos with a single or multiple aneuploidy displayed temporal morphokinetic variables that were significantly different from euploid embryos. If any variants were established, a further objective was to develop a model to categorize the risk of aneuploidy in embryos based on this non-invasive morphokinetic data.
Data obtained for this research were obtained from the treatment of 25 couples attending an independent IVF clinic (CARE Fertility, Manchester, United Kingdom) from May 2011 to July 2012. All protocols complied with UK regulation (Human Fertilisation and Embryology Act, 1990, 2008). The study did not require ethical or institutional review board approval, having been performed according to previously validated procedures. This was a retrospective cohort study blinded to ploidy, with recording of embryo development using time-lapse technology (EmbryoScope; Unisense Fertilitech, Denmark) and strict adherence to annotation protocols for the variables defined in Table 1.
The couples selected for this study requested or were recommended PGS due to a history of recurrent implantation failure following IVF (defined as more than two failed IVF attempts), recurrent miscarriage (defined as more than two spontaneous miscarriages), severe male factor infertility, previous aneuploidy or advanced female age (defined as >37![]() years). Female age ranged from 31 to 47
years). Female age ranged from 31 to 47![]() years (mean
years (mean![]() ±
±![]() standard deviation 38.6
standard deviation 38.6![]() ±
±![]() 3.6
3.6![]() years).
years).
Pituitary suppression and ovarian stimulation was performed in 75% of patients with a gonadotrophin-releasing hormone agonist (Suprecur; 0.5![]() ml subcutaneously daily; Sanofi Aventis, UK) or antagonist for the remainder (Cetrotide; 0.25
ml subcutaneously daily; Sanofi Aventis, UK) or antagonist for the remainder (Cetrotide; 0.25![]() mg daily; Merck Serono, UK). Ovarian stimulation was achieved using human menopausal gonadotophin (Menopur, Ferring, UK) and/or recombinant FSH (Gonal-F; Merck Serono), with doses ranging from 150 to 600
mg daily; Merck Serono, UK). Ovarian stimulation was achieved using human menopausal gonadotophin (Menopur, Ferring, UK) and/or recombinant FSH (Gonal-F; Merck Serono), with doses ranging from 150 to 600![]() IU per day according to patient type and response. No differences were observed in ploidy for the stimulation regimens or doses (data not shown).
IU per day according to patient type and response. No differences were observed in ploidy for the stimulation regimens or doses (data not shown).
With the female patient under sedation with a combination of propofol (Braun, Germany), fentanyl (Auden McKenzie, UK) and midazolam (Hamelyn, UK), transvaginal ultrasound-guided oocyte retrieval took place 36![]() h post human chorionic gonadotrophin injection (10,000
h post human chorionic gonadotrophin injection (10,000![]() IU; Pregnyl; Organon, UK; or Ovitrelle; Merck Serono) or agonist trigger, using a dual lumen aspiration needle (Swemed; Vitrolife, UK) connected to a vacuum pump (Rocket Medical, UK). Oocyte–cumulus-complexes were recovered from follicular aspirates using a stereomicroscope in a class II hood with a heated stage, washed and cultured in Ferticult IVF medium (Fertipro, Belgium) at 5% carbon dioxide in air, 37.0°C, maximum humidity, for between 2 and 4
IU; Pregnyl; Organon, UK; or Ovitrelle; Merck Serono) or agonist trigger, using a dual lumen aspiration needle (Swemed; Vitrolife, UK) connected to a vacuum pump (Rocket Medical, UK). Oocyte–cumulus-complexes were recovered from follicular aspirates using a stereomicroscope in a class II hood with a heated stage, washed and cultured in Ferticult IVF medium (Fertipro, Belgium) at 5% carbon dioxide in air, 37.0°C, maximum humidity, for between 2 and 4![]() h before cumulus cell denudation with 15–20
h before cumulus cell denudation with 15–20![]() IU/ml cumulase (Origio, Denmark) in the same medium and complete removal of the coronae radiatae with a 140
IU/ml cumulase (Origio, Denmark) in the same medium and complete removal of the coronae radiatae with a 140![]() μm pipette (EZ Squeeze; Research Instruments, UK). Oocytes at the metaphase-II stage underwent insemination by intracytoplasmic sperm injection (ICSI).
μm pipette (EZ Squeeze; Research Instruments, UK). Oocytes at the metaphase-II stage underwent insemination by intracytoplasmic sperm injection (ICSI).
Following ICSI, oocytes were placed individually in microwells of EmbryoSlides (Unisense Fertilitech) in 25![]() μl Global IVF medium (LifeGlobal) supplemented with 10% dextran serum supplement (Irvine Scientific) and were overlaid with 1.4
μl Global IVF medium (LifeGlobal) supplemented with 10% dextran serum supplement (Irvine Scientific) and were overlaid with 1.4![]() ml mineral oil (Fertipro, Belgium) in the EmbryoScope. EmbryoSlides were prepared around the time of oocyte recovery with medium and oil that had equilibrated overnight.
ml mineral oil (Fertipro, Belgium) in the EmbryoScope. EmbryoSlides were prepared around the time of oocyte recovery with medium and oil that had equilibrated overnight.
Once loaded with the inseminated oocytes, EmbryoSlides were placed into the EmbryoScope time-lapse incubator at 37C in 5.5% CO2, 5% O2 and 89.5% N2 for at least 5![]() days. Culture was temporarily interrupted on day 3 in order to re-fresh the medium. This was performed by removing 20
days. Culture was temporarily interrupted on day 3 in order to re-fresh the medium. This was performed by removing 20![]() μl of culture medium from each microwell and replacing it with fresh, pre-equilibrated medium. The built-in microscope was used to acquire images of each fertilized oocyte every 20
μl of culture medium from each microwell and replacing it with fresh, pre-equilibrated medium. The built-in microscope was used to acquire images of each fertilized oocyte every 20![]() min through seven focal planes.
min through seven focal planes.
Of the blastocysts in this study, 69 (70.41%) of them underwent a series of three 4-μm diameter laser pulses (Saturn Active laser and Integra micromanipulator; Research Instruments) to breach the zona pellucida on day 3 of culture in order to facilitate trophectoderm herniation for biopsy. For the remaining 29 (29.59%) blastocysts, laser breaching was performed at the time of the biopsy procedure. No differences were observed in time from insemination to start of blastulation (tSB) or time from insemination to formation of a full blastocyst (tB) between embryos which underwent this procedure and those which did not.
The timing of the procedure and the stage of blastocyst development when trophectoderm biopsy was undertaken varied in this study as they were performed in a minimum number of cohorts to reduce disruption to the incubation and for efficient working practice. All biopsies were performed on blastocyst-stage embryos on day 5 or 6 post oocyte recovery. The minimum stage and quality criteria for selection of blastocysts for biopsy and vitrification in this study were embryos that were at least full blastocysts, with at least a fair, easily discernible inner cell mass and at least a few cells forming a loose trophectoderm (equivalent to stage 2 grade 2; Alpha and ESHRE, 2011a, Alpha and ESHRE, 2011b).
The time of hatching could not be reliably assessed in the embryos where a facilitative breach was made in the zona pellucida. Where observed, the time from insemination to start of blastulation was recorded for the first expansion episode only and only when expansion occurred prior to biopsy. The cycles of collapse and expansion, although observed in some of the blastocysts, were not recorded.
Up to three blastocysts per patient were biopsied in one dish in individually labelled 20-μl microdrops of G-MOPS buffered medium (Vitrolife, Sweden) using a 33–37-μm inner diameter blastomere biopsy micropipette (Humagen, Origio). Where required, the zona pellucida was breached using a series of 4-μm laser pulses and 5–10 trophoblast cells aspirated into the biopsy pipette. Stronger laser pulses of up to 15![]() μm were used with aspiration and mechanical pressure to remove the cells from the blastocyst. In compliance with UK regulation and good practice, witnessing was performed at every stage by manual witnessing or Matcher barcode electronic witnessing (IMT International). Time-lapse monitoring ceased for the particular blastocysts that were removed from the EmbryoScope for biopsy. Following biopsy, the biopsied cells were placed into 0.2
μm were used with aspiration and mechanical pressure to remove the cells from the blastocyst. In compliance with UK regulation and good practice, witnessing was performed at every stage by manual witnessing or Matcher barcode electronic witnessing (IMT International). Time-lapse monitoring ceased for the particular blastocysts that were removed from the EmbryoScope for biopsy. Following biopsy, the biopsied cells were placed into 0.2![]() ml thin walled-tubes and sealed and frozen by placing them into a freezer at −20°C prior to genetic screening and the blastocysts vitrified (Kitazato; Hunter Scientific, UK).
ml thin walled-tubes and sealed and frozen by placing them into a freezer at −20°C prior to genetic screening and the blastocysts vitrified (Kitazato; Hunter Scientific, UK).
Trophectoderm biopsies of 98 blastocysts were amplified and analysed by either whole-genome amplification and array comparative genomic hybridization (n![]() =
=![]() 37; Genesis Genetics Europe, Nottingham, UK) or by single-nucleotide polymorphism microarray (n
37; Genesis Genetics Europe, Nottingham, UK) or by single-nucleotide polymorphism microarray (n![]() =
=![]() 61; Natera, California, USA). Embryos were categorized as ‘single’ or ‘multiple’ aneuploid depending on the number of chromosomes affected.
61; Natera, California, USA). Embryos were categorized as ‘single’ or ‘multiple’ aneuploid depending on the number of chromosomes affected.
The method by which analysis of trophectoderm biopsies via single-nucleotide polymorphism microarray occurred has previously been described (Johnson et al., 2010a, Johnson et al., 2010b). Briefly, biopsies were lysed and heat inactivated prior to DNA amplification. In order to determine the source of aneuploidy, parental buccal swabs or blood samples were collected. Genomic DNA was isolated and the standard Infinium II protocol (Illumina, San Diego, CA, USA) was used for analysis of these samples. The genotype of the biopsy samples was determined using Infinium II genotyping microarrays (CytoSNP-12 chips). Chromosome imbalances were determined using a specific algorithm, described in Johnson et al. (2010b), which used the information gained from the parental genotype and the biopsy sample to determine both whole-chromosome imbalances and structural abnormalities, along with the parental source of the aberration.
The method by which analysis of trophectoderm biopsies via array comparative genomic hybridization occurred as has previously been described (Fishel et al., 2011). Briefly, DNA was amplified from the biopsied cells using the SurePlex DNA Amplification System (BlueGnome, Cambridge, UK) according to the manufacturer’s instructions. Amplification was assessed by DNA gel electrophoresis and only samples generating amplified product were labelled. Test and control sample product and SureRef Male DNA (BlueGnome) were labelled with Cy3 and Cy5, respectively, using the BlueGnome fluorescent labelling system, according to the manufacturer’s instructions. Labelled test and control sample and SureRef Male DNA were co-hybridized using 24Sure microarrays version 2 (BlueGnome). The resulting 24Sure microarrays were hybridized, washed and scanned according to the manufacturer’s protocol. Scanned images were analysed and quantified and whole-chromosomal copy number ratios were reported using the Cytochip algorithm fixed settings in BlueFuse Multi Software (BlueGnome).
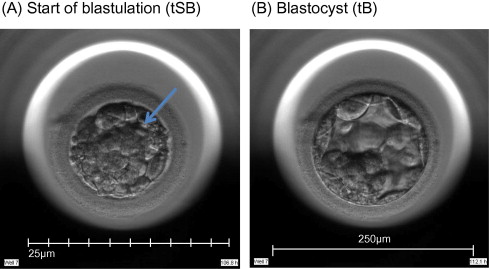
Time-lapse images were collected for the duration of the culture period, to the point of biopsy and were used for the assessment of fertilization and embryo development. The time of insemination by ICSI was programmed into the EmbryoScope when the slide was loaded, as the time point midway through the ICSI procedure. The EmbryoViewer image analysis software was used to log and display the precise timing of developmental events as they were annotated by the embryologists studying the time-lapse images. Table 1 outlines the definitions of morphokinetic variables that were recorded in this study of 2 pronuclei embryos up to the point of trophectoderm biopsy. All times were recorded in hours post insemination (hpi). All annotations were made prior to the result of the biopsy analysis and were therefore blind. Example images show how the start and completion of blastulation were identified (Figure 1).
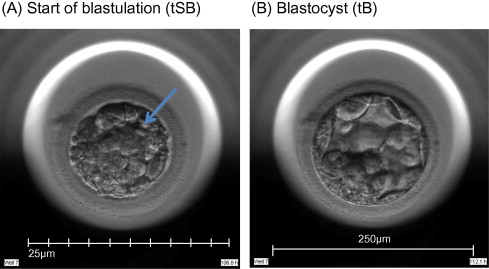
Time-lapse images of the same embryo demonstrating its appearance at specific time points as utilized in this study. (A) Human embryo at 106.8![]() hpi, with the start of a cavity forming (arrow), at tSB. (B) Human blastocyst at 112.1
hpi, with the start of a cavity forming (arrow), at tSB. (B) Human blastocyst at 112.1![]() hpi, with a blastocoele filling the embryo with <10% increase in its diameter, at tB.
hpi, with a blastocoele filling the embryo with <10% increase in its diameter, at tB.
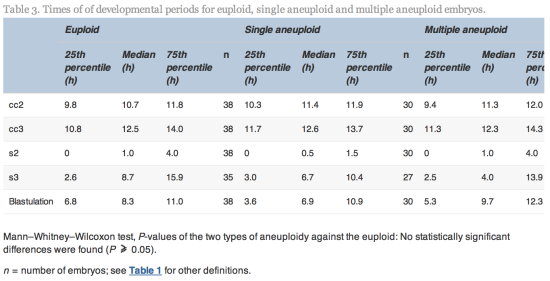
Embryos with incomplete annotations, failed amplification and/or abnormal or lack of fertilization were excluded from analysis. The time variables were tested for normality using Shapiro–Wilks test for normality. Since most variables were found not to be normally distributed, non-parametric tests were used to determine whether differences were significant. The Mann–Whitney–Wilcoxon test was used to test differences in morphokinetic variables, whilst Fisher’s test (odds ratio different to one) was used to test differences in incidence rate of multinuclearity and irregular cleavage patterns. The statistical analyses were performed using R statistical software version 2.15.0 (R Foundation for Statistical Computing).
Morphokinetic variables found to differ between euploid and aneuploid embryos (single and multiple aneuploid combined) were used to build a decision tree model by recursive partitioning to partition the embryos into groups depending on the value of the variables tSB and tB. The recursive partitioning was optimized by the logWorth value. The partitioning process stopped when all new steps had a logWorth value below one (non-significant splits). The model classified embryos into three classes of aneuploidy risk: low, medium or high risk. The recursive partitioning and the probability of a random embryo in a particular risk class being aneuploid were calculated in JMP version 10.0 (SAS Institute).
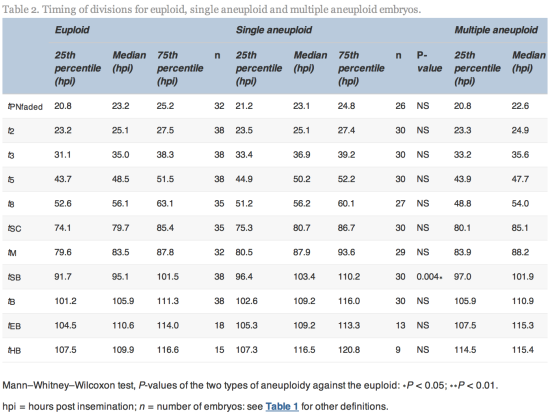
The time of initiation of compaction (tSC) was significantly delayed (P![]() =
=![]() 0.02) for multiple aneuploid embryos (median 85.1
0.02) for multiple aneuploid embryos (median 85.1![]() hpi, range 64.9–113.0
hpi, range 64.9–113.0![]() hpi) compared with euploid embryos (median 79.7
hpi) compared with euploid embryos (median 79.7![]() hpi, range 56.3–107.6
hpi, range 56.3–107.6![]() hpi). The time of initiation of blastulation (tSB) was significantly delayed (P
hpi). The time of initiation of blastulation (tSB) was significantly delayed (P![]() =
=![]() 0.004 and 0.006) more than 6 h for both single aneuploid embryos (median 103.4, range 79.8–121.5
0.004 and 0.006) more than 6 h for both single aneuploid embryos (median 103.4, range 79.8–121.5![]() hpi) and multiple aneuploid embryos (median 101.9
hpi) and multiple aneuploid embryos (median 101.9![]() hpi, range 86.8–129.4
hpi, range 86.8–129.4![]() hpi) compared with euploid embryos (median 95.1
hpi) compared with euploid embryos (median 95.1![]() hpi, range 85.2–113.9
hpi, range 85.2–113.9![]() hpi). The time of full blastulation (tB) was significantly delayed (P
hpi). The time of full blastulation (tB) was significantly delayed (P![]() =
=![]() 0.01) for 5
0.01) for 5![]() h for multiple aneuploid embryos (median 110.9
h for multiple aneuploid embryos (median 110.9![]() hpi, range 90.1–137.0
hpi, range 90.1–137.0![]() hpi) compared with euploid embryos (median 105.9, range 86.8–122.3
hpi) compared with euploid embryos (median 105.9, range 86.8–122.3![]() hpi). All other timings tested were not significantly different (Table 2).
hpi). All other timings tested were not significantly different (Table 2).
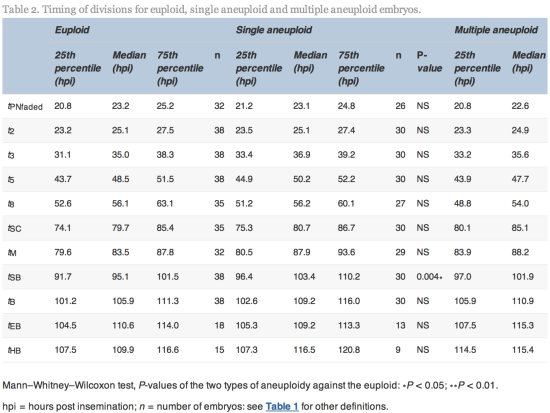
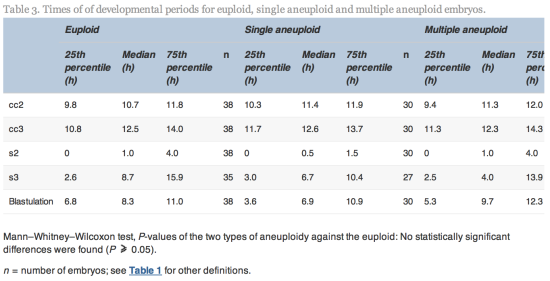
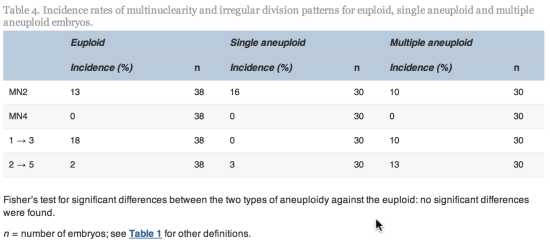
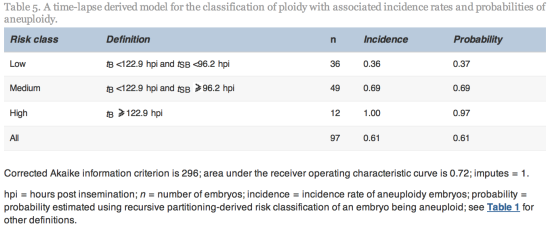
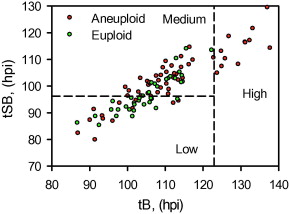
Since tSB and tB were found to differ significantly between euploid and aneuploidy embryos, these variables were selected for the simple classification model with three risk classes of aneuploidy (Table 5 and Figure 2).
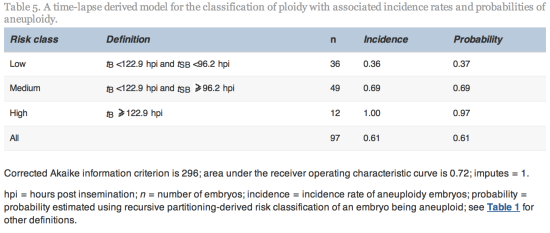
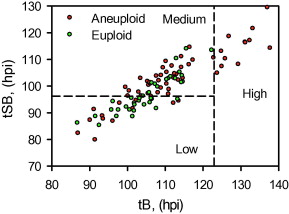
Figure 2
The three classes of aneuploidy risk (low, medium and high) based on time from insemination to a full blastocoele where the blastocyst has not yet started expansion (tB) and time from insemination to start of blastulation (tSB). Area under the receiver operating characteristic curve is 0.72.![]() hpi
hpi![]() =
=![]() hours post insemination.
hours post insemination.

As far as is known, this is the first paper demonstrating an association between human embryo ploidy and morphokinetics derived from time-lapse technology. The recent retrospective study by Meseguer et al. (2012), which set out to quantify the effect of using stable time-lapse culture conditions and morphokinetic variables for embryo selection, used clinical pregnancy as the end point. This was defined by the presence of gestational sacs with fetal heartbeat during week 7 of gestation. However, it is likely that a proportion of these early IVF pregnancies will be aneuploid. Fragouli and Wells (2011) discussed the landscape of blastocyst comprehensive cytogenetic analysis and reported overall aneuploidy rates in blastocysts to be greater than 50%. They suggest that the majority of biological selection against aneuploidy and genetic anomalies occurs at or after the time of implantation. It is widely accepted that a major cause of failed implantations or miscarriage following embryo transfer is the (unknowing) transfer of aneuploid embryos. Up until now, the only means available to IVF centres for assessing ploidy in embryos has been polar body, blastomere or trophoblast biopsy with PGS. Ploidy of the embryos in this study was assessed by screening of the trophectoderm to give full chromosome copy number, which is considered to give high concordance with the inner cell mass, but may still not be 100% accurate.
Alfarawati et al. (2011) previously compared blastocyst qualitative morphology with ploidy and demonstrated only a weak association between blastocyst morphology and aneuploidy. They reported that, concerning the growth rate of blastocysts, there was an insignificant trend toward aneuploid embryos showing slower progression to the most advanced blastocyst stages and that embryos with complex aneuploidy were most delayed. This study was performed without the benefit of time-lapse technology and the findings were not clinically applicable as no clear cut-off time point was given for discrimination between complex aneuploid embryos and euploid ones. The time or frequency of blastocyst morphological assessment was not discussed in that paper.
From the cohort of blastocysts that underwent PGS in this study, the overall incidence rate of aneuploidy was 61%. The model used the morphokinetic variables tSB and tB of these embryos of known ploidy to classify a blastocyst’s risk of aneuploidy. The model can therefore be used to rank individual and unscreened blastocysts as having low (probability 0.37), medium (probability 0.69) or high risk (probability 0.97) of aneuploidy. Especially where there is a choice of embryos available, the model could be useful in reducing the chance of selecting an aneuploid embryo. The classification from this model may, therefore, be used clinically to rank embryos for transfer and could also be used alongside PGS in order to prioritize embryos for biopsy and screening, particularly where costs are based on the number of embryos screened. Additional data may facilitate the fine tuning of this algorithm.
Embryos with rapid cleavage from 2 to 3 cells in less than 5![]() h have been reported to have a significantly lower implantation potential than embryos with a normal cell cycle length (Rubio et al., 2012). The normal cell cycle time has been established to be 10–12
h have been reported to have a significantly lower implantation potential than embryos with a normal cell cycle length (Rubio et al., 2012). The normal cell cycle time has been established to be 10–12![]() h (Cummins et al., 1986) and therefore there is sufficient time for DNA replication. It is unknown if a causal relationship to ploidy exists in all cases of such anomalous cleavage: whether rapid cleavage provides insufficient time for complete genome replication resulting in aneuploidy or whether a particular existing aneuploidy results in erroneous cleavage patterning. For example, some aneuploidies are lethal during the preimplantation stage, such as many monosomies, whilst some, such as trisomies 13, 18 and 21, are compatible with full-term delivery. Somfai et al. (2010)reported a high frequency of chromosomal anomalies in bovine embryos which developed from zygotes that exhibited direct division to three from one cell, although rapid cleavage was observed in a small proportion of both euploid (18%) and aneuploid embryos (5% overall) in the present study. The incidence of such rapid cleavage was not assessed in embryos with arrested development in this study and further research into such cleavage patterning, such as reverse cleavage, is underway.
h (Cummins et al., 1986) and therefore there is sufficient time for DNA replication. It is unknown if a causal relationship to ploidy exists in all cases of such anomalous cleavage: whether rapid cleavage provides insufficient time for complete genome replication resulting in aneuploidy or whether a particular existing aneuploidy results in erroneous cleavage patterning. For example, some aneuploidies are lethal during the preimplantation stage, such as many monosomies, whilst some, such as trisomies 13, 18 and 21, are compatible with full-term delivery. Somfai et al. (2010)reported a high frequency of chromosomal anomalies in bovine embryos which developed from zygotes that exhibited direct division to three from one cell, although rapid cleavage was observed in a small proportion of both euploid (18%) and aneuploid embryos (5% overall) in the present study. The incidence of such rapid cleavage was not assessed in embryos with arrested development in this study and further research into such cleavage patterning, such as reverse cleavage, is underway.
This study developed a model based on this study centre’s own culture systems, in which all controllable environmental factors have been standardized and with a strict annotation policy followed by laboratory staff. This generated two precise time points, the start of blastulation and the time that the embryo reached the full blastocyst stage, as important markers in this model for classifying the risk of ploidy. With standard incubation, embryologists will be unable to annotate these timings precisely without tremendous disruption and compromise to incubation and working practice. Whilst a daily observational procedure could be applied on day 4 at around the same time of day as the insemination occurred (∼96![]() hpi) and another on day 5 at a similar time (120
hpi) and another on day 5 at a similar time (120![]() hpi) in an attempt to align with the temporal values in this model, the facility to study and rewind time-lapse images to identify the point of blastulation initiation (tSB) and completion of blastulation (tB) is essential to this modelling to permit a high degree of confidence in this data. Furthermore, the accuracy of the model is dependent on the 20-min imaging interval (i.e. capable of generating 19 images in 6
hpi) in an attempt to align with the temporal values in this model, the facility to study and rewind time-lapse images to identify the point of blastulation initiation (tSB) and completion of blastulation (tB) is essential to this modelling to permit a high degree of confidence in this data. Furthermore, the accuracy of the model is dependent on the 20-min imaging interval (i.e. capable of generating 19 images in 6![]() h) It is unknown at this time whether specific time points from this model can be applied to a different culture system, culture media, plastic ware, gas mix, air purity, etc., even with time-lapse technology.
h) It is unknown at this time whether specific time points from this model can be applied to a different culture system, culture media, plastic ware, gas mix, air purity, etc., even with time-lapse technology.
This model for categorizing the risk of aneuploidy using non-invasive methods should improve clinical outcome. Other systems that simply predict the occurrence of an embryo to undergo blastulation (Wong et al., 2010) are unlikely to be as clinically robust given that all aneuploid pregnancies, miscarriages and live births, ipso facto, developed to and beyond the blastocyst stage, as did the many aneuploid blastocysts that failed to become pregnancies. The high incidence of aneuploidy in blastocysts (61% in this study), confirms the clinical limitations of time-lapse systems used only to select embryos that are predicted to become blastocysts.
The period leading up to blastulation is a period of intense cellular metabolic activity, gene activation, rapidly increasing cell division and differentiation. Cell division and the mitotic process is a series of complex structural rearrangements involving the kinetochore attachments to the microtubules (Gonen et al., 2012), cohesion molecules for the crucially precise separation of the chromosome to ensure correct alignment on the spindle (Clift and Marston, 2011), the spindle assembly complex and many highly specialized proteins subject to precise gene expression (Vogt et al., 2008). Although the cause of a temporal delay in aneuploid embryos compared with their euploid counterparts is not yet fully explained, there exist error detection and repair systems within the cell to prevent aneuploidy (Nasmyth and Haering, 2009). It is highly probable, therefore, that mitotic errors in individual cells at this stage of the rapidly developing embryo involve complex biochemical systems delaying karyo- and cytokinesis, which result in the gross observation of delayed blastulation.
Time-lapse photography in a closed incubation system such as the EmbryoScope, when used precisely to measure the initiation and completion of blastulation, appears to be the most reliable non-invasive method of ranking the risk of aneuploidy in advanced-stage embryos. It is unlikely ever to be as absolute as gaining accurate chromosome copy number from biopsied cells, but it is an important tool to enhance the chances of a live birth following IVF by non-invasive means. Further studies are underway to quantify further morphokinetic variables, to observe the impact of specific aneuploidy data and to prospectively test this model.
This study indicates that, with time-lapse monitoring of embryo development to blastocyst in a closed incubation system, it is possible, where there are alternative embryos available within a cohort, to avoid the selection of embryos having a high risk of aneuploidy and to preferentially select embryos with a greatly reduced risk of aneuploidy based on morphokinetic timing.
This non-invasive approach may be offered to patients as an alternative to PGS or, indeed, as a complementary system. This model for classifying the risk of aneuploidy requires time-lapse technology to enable identification of specific time points for embryo development up to the blastocyst stage. This accessible and non-invasive embryo selection model may be used for patients electing against invasive genetic screening technology or for clinics without the skills or access to PGS.
The authors wish to thank Mette Laegdsmand MSc, PhD for data mining and statistical analysis support. They also thank the CARE Fertility Manchester team for their enthusiasm and support.
http://www.rbmojournal.com/article/S1472-6483(13)00070-9/fulltext

 (3)
(3)
6840